We develop and implement genome engineering approaches to systematically connect cancer genotypes to phenotypes with the ultimate goal to translate the obtained knowledge into more effective therapies for currently difficult to treat cancers.
For that purpose we employ genome engineering technologies that allow us to (1) reversely engineer cancer mutations into the genome of cultured cells, (2) to activate the transcription of cancer genes from their endogenous genomic loci and (3) to inhibit their expression by targeted degradation of their transcripts.
Figure 1 explains the concepts behind those cutting-edge CRISPR technologies.
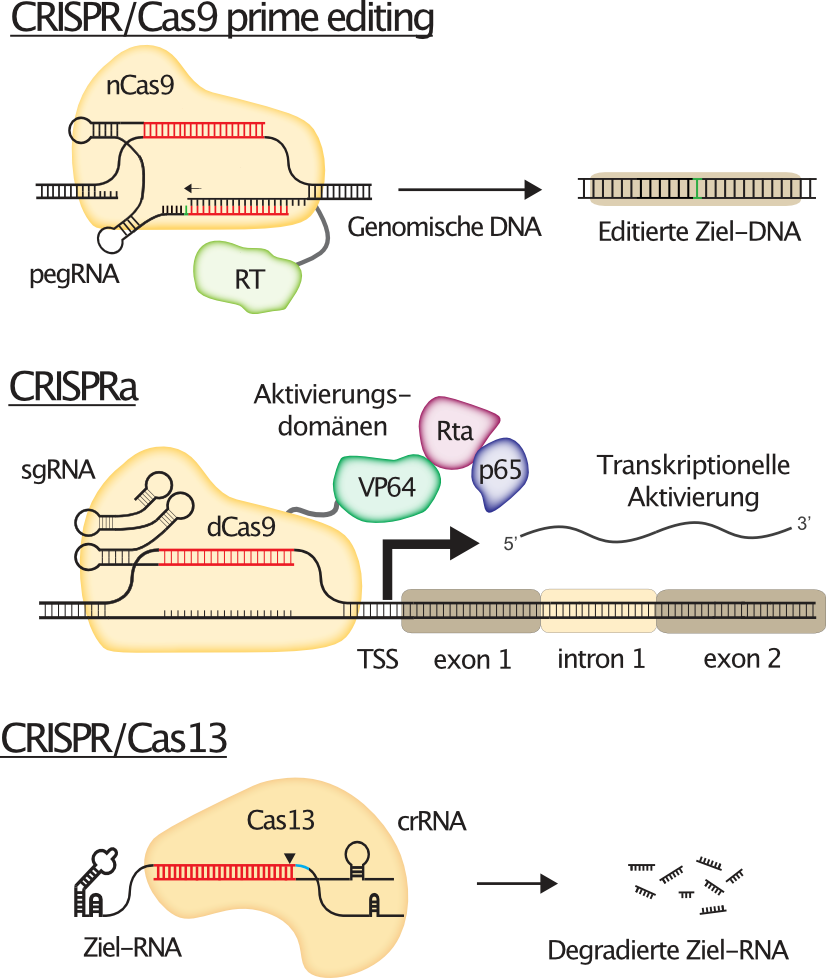
While CRISPR/Cas9 nuclease from S.pyogenes was the first CRISPR system that was successfully used to edit human cells, it by far did not remain the only one. Adaptations of the CRISPR/Cas9 system now allow the precise modification of specific target sites in the human genome, most notably via prime editing. Furthermore, catalytically inactive or dead versions of Cas9 (dCas9) are used as programmable "homing devices" to target functional domains to almost any destination in the genome, for instance to allow the transcriptional activation of target genes from their endogenous genomic loci (CRISPRa). But also CRISPR systems other than CRISPR/Cas9 have found their way into the human genome engineering toolbox, such as the RNA targeting Cas13 for instance, a ribonuclease that can be used to specifically degrade any target RNA of interest, thereby facilitating reversible gene silencing.
Since the individual investigation of cancer associated genetic and epigenetic alterations would be extremely time consuming, we develop scalable gene perturbation approaches, so called pooled genetic screens, which allow the analysis of hundreds of thousands of perturbations in a single reaction vessel (Figure 2). Moreover, we combine different CRISPR systems to introduce orthogonal perturbations in the same cell, an approach that allows us to infer the direction of the flow of information in genetic networks.
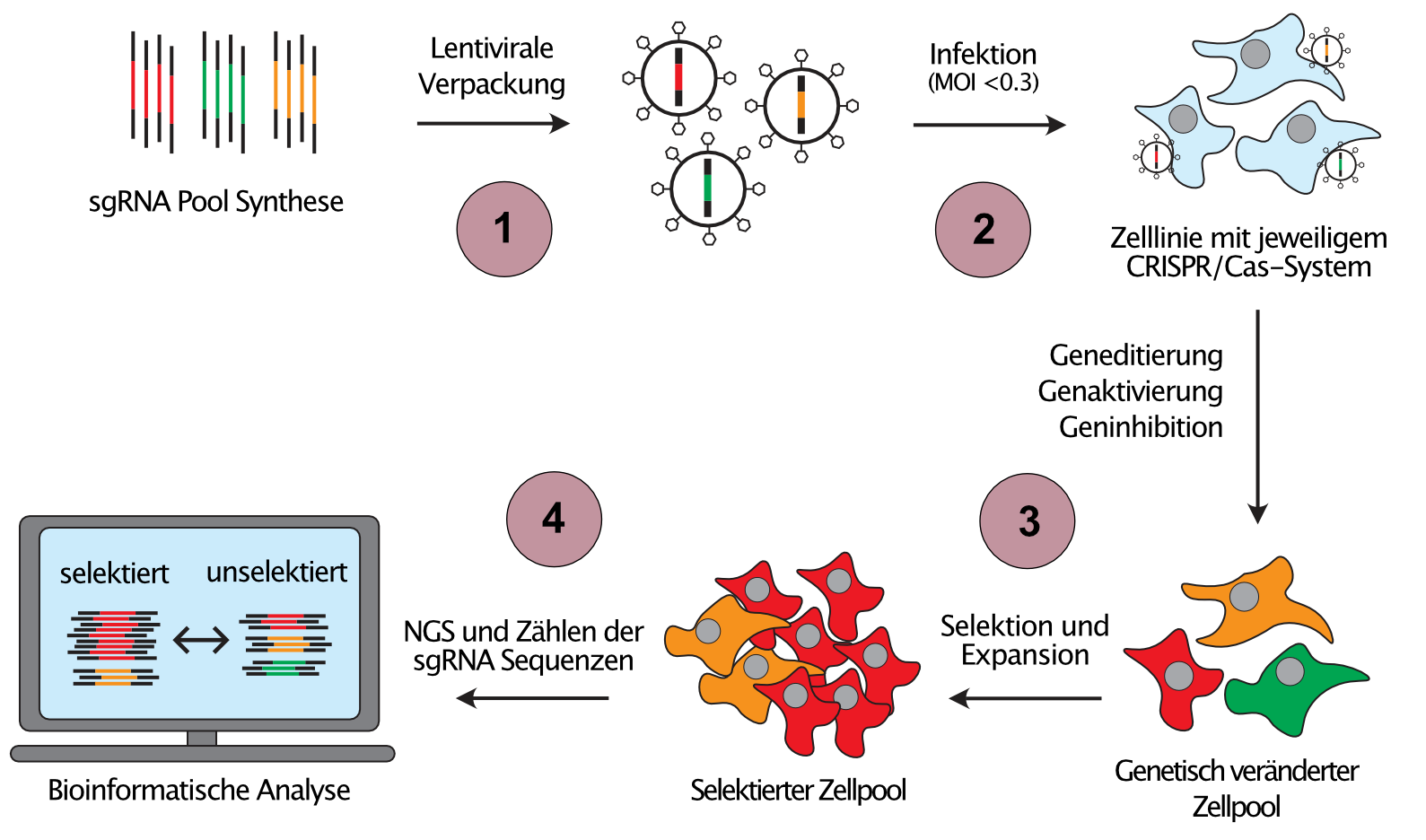
(1) Library pools of sgRNA template sequences targeting the regions of interest are synthesized and cloned into sgRNA expression vectors. (2) Using lentiviral transduction at low MOI ensures the expression of maximum one sgRNA per target cell and consequently, a unique perturbation is introduced into every target cell. (3) Cell pools are then challenged with the selection of interest, e.g. proliferation, a drug or a phenotype that is sortable by flow cytometry. Cells with perturbations that cause resistance against the selection pressure will enrich (red cells), while cells with sensitising perturbations will be depleted from the selected cell pool (green cells). (4) Next-Generation-Sequencing (NGS) and read counting of the sgRNA sequences before and after selection allows the identification of those perturbations that caused resistance or sensitivity.
In addition to the phenotypic screens outlined in Figure 2 we employ CRISPR screens with single cell transcriptome read out to study the deregulation of transcriptional networks in cells with engineered cancer associated alterations (Figure 3). We are currently optimizing the methodology for these types of screens as part of the PeCaN consortium, which is aimed at the parameterisation of computational models for the development of personalised therapeutic strategies for patients with advanced triple negative breast cancer (TNBC), a particularly aggressive and difficult to treat form of breast cancer.
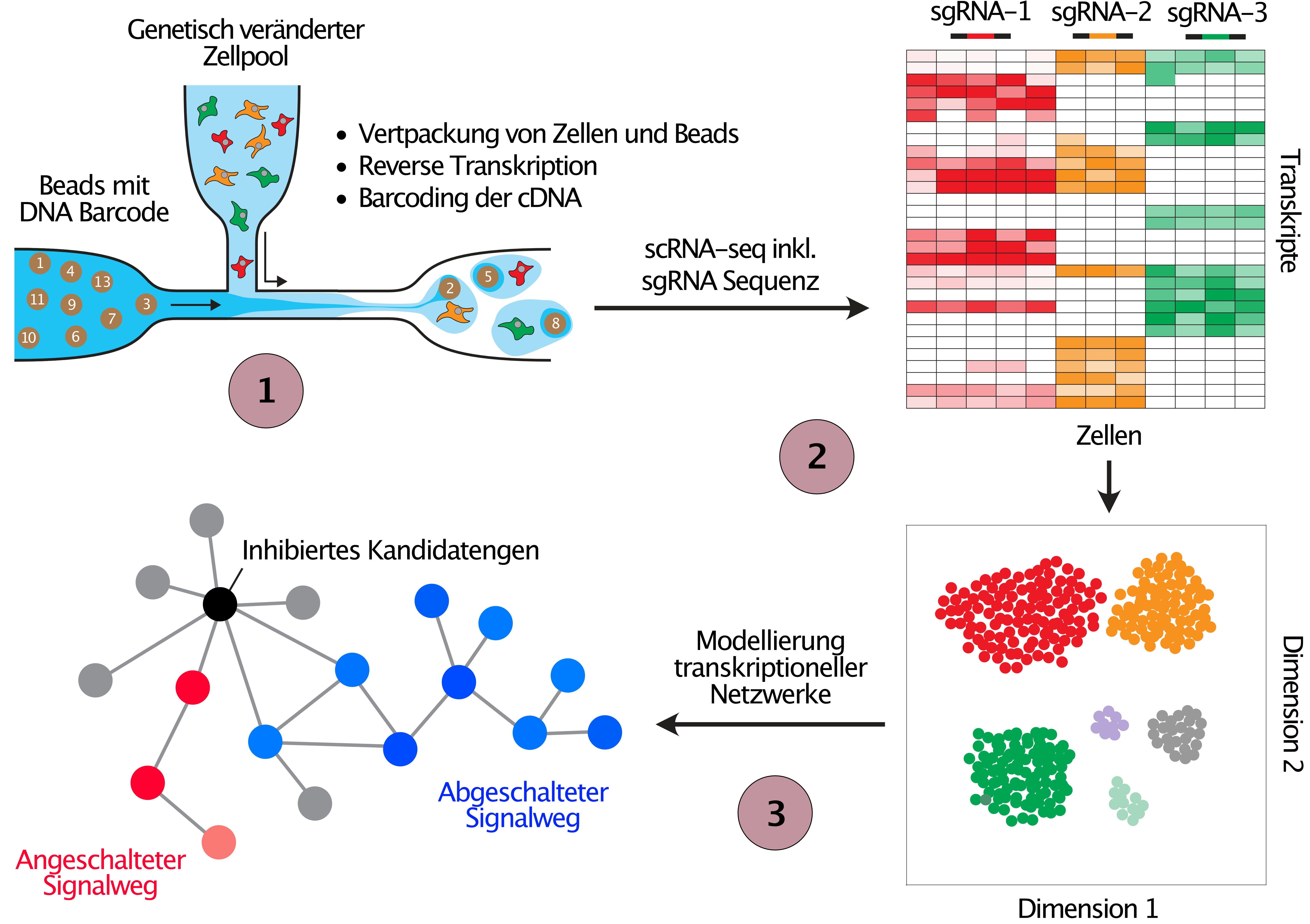
(1) Cell pools are perturbed exactly as explained in Figure 2, before they are run through the 10X Genomics single cell gene expression pipeline. In parallel to the transcriptome, the sequence of the expressed sgRNA is detected in every single cell, and hence the perturbed target can be connected to resulting gene expression changes in each cell. (2) Cells are clustered by the similarity of their gene expression profiles (t-SNE) where similar changes in gene expression following the perturbation of genes suggests similar function. (3) The obtained data from gene expression changes after candidate gene perturbation will be used to develop mathematical models to understand the gene expression networks that are deregulated in cancer patients with the tested genetic perturbation.
Funding
2022 - 2025: German Research Foundation - GRK 2751 InCuPANC
2021 - 2024: Medical Faculty of the Martin Luther University - Wilhelm-Roux-Program
2020 - 2023: Bayer AG
2020 - 2023: Federal Ministry of Education and Research - PeCaN
2019 - 2022: European Social Fund (ESF) - HAL-OX